
Rapid Whole Genome Sequencing and Analysis Using AI Emedgene Poster
Whole genome sequencing (WGS) has become a standard method in clinical diagnostic laboratories, where it represents a reliable analytical tool due to its excellent coverage quality, enabling deep insights into the genome. The PCR-free protocol offers a superior solution for traditionally difficult-to-sequence DNA regions, such as GC-rich regions, promoters, and repetitive content, while also reducing library bias and gaps. In addition, WGS can be used to make accurate diagnoses of very rare disorders that would otherwise require harmful and invasive diagnostic procedures. This protocol enables the rapid preparation of high-quality, sequencing-ready indexed whole-genome sequencing libraries, sequencing, and bioinformatic analysis within 40 hours at a low cost.

Rapid whole-genome sequencing and comprehensive analysis using AI Emedgene
Purpose: Protocol optimization of the rapid whole-genome sequencing providing highquality data with subsequent annotation of priority
variants using the AI software tool Emedgene.
Materials and methods: Genomic DNA was isolated using the EZ1&2 DNA Blood 350 μl Kit from venous blood from 3 participants (family
trio: mother, father and proband daughter); proband diagnosed with Mayer-Rokitansky-Küster-Hauser syndrome. Whole-genome libraries
were prepared using the TruSeq DNA PCR-Free High Throughput Library Prep Kit. Sequencing was performed on a NovaSeq 6000 using S1
flowcell sequencing chemistry. Dynamic Read Analysis for GENomics (DRAGEN) On Premise Server v4.1 (Illumina) with hg38 Alignment and
Variant Calling and DRAGEN v4.0 Cloud System and AI software tool Emedgene (Illumina) were used for subsequent secondary analysis.
Results: High-quality genomic DNA was successfully isolated from the venous blood of all 3 study participants. Whole-genome libraries
were successfully sequenced with a generated volume of data: 464.92 Gb, with 92.57% of reads covered more than 35× and 94.66% of
reads with a Phred quality score above 30 (Q30). All whole-genome libraries met the required criterion of 800 million of pair-end reads.
Secondary analysis using the DRAGEN v4.0 cloud system and artificial intelligence Emedgene identified 7 pathological variants of Mayer-
Rokitansky-Küster-Hauser syndrome, which were evaluated by a laboratory diagnostician.
Conclusion: The optimized protocol enables rapid preparation of high-quality indexed wholegenome sequencing libraries, sequencing
and bioinformatic analysis in 39 hours and 54 minutes.
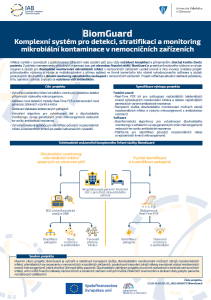
BiomGuard - Comprehensive System for microbial contamination management in hospitals
Infections arising in connection with the provided health or social care are always an undesirable complication and at the very least impair the quality of the patient ´ s life. For management of the respective organizations, it represents a huge financial burden too. BiomGuard is an industrial research and development project providing kits and services for long-term monitoring of microbial infections in hospital facilities of a high level and degree of innovation. The project is based on multidisciplinary approach with direct application in the form of a commercial kit including evaluation software and services providing long-term and detailed monitoring of microbial representation in hospital facilities. The project reflects current urgent requirements market, especially due to increasing resistance to antibiotics.

DiPha - a tool for prediction and guidance of purposeful pharmacotherapy
Medical doctors currently prescribe a number of medicinal products, while scientific studies describe that the drugs are effective in only 60% of patients and also a significant number of patients are hospitalized for the occurrence of serious side effects after applied pharmacotherapy. Genetic factors, i.e. information stored in our DNA, are proven to play a key role in the reaction to drugs. Pharmacogenomics is a field that studies the influence of the patient's genetics predisposition to the drug's effectiveness and, on the other hand, to the occurrence of adverse effects. The result of the pharmacogenetic examination affects the choice of a drugs dosage according to the genetic profile of individual patients. With the significantly increasing number of prescribed drugs, the need to boost the capacity of examinations that determine the genetic factors influencing the effects of drugs is also increasing, as weel as requirement to improve the implementation of the results of these examinations into clinical practice leading to correct treatment decisions. A possible way to improve accessibility is the screening genetic testing, when collected comprehensive pharmacogenomic information and are readily available for future drug prescribing decisions.

Centre for Digital Genomics 23 - CeDiGE 23
The Institute of Applied Biotechnologies (IAB) is pleased to announce the successful completion and implementation of its new Infrastructure, starting the Centre for Digital Genomics. This new complex of hardware and software systems is specifically designed to support the growing demand for next-generation sequencing (NGS) in diagnostics, particularly in areas such as oncology, metabolic disorders, and prenatal testing.
Developed LIMS system will automate complex sample handling processes, allowing clients to reach faster, more reliable, and cost-effective sequencing solutions. Installed hardware infrastructure will provide capacity of storage and processing of data even for the largest projects. The implemented system ensures better management of large genomic datasets, reduces human error, and streamlines the analysis of sequencing data.
The IAB team is excited to start using the new system to enhance its services, providing quicker results while maintaining high standards of quality, essential for personalized medicine and advanced genetic research.

Automation of High-Throughput Whole Exome Library Preparation
Whole-exome sequencing (WES) is a powerful clinical tool for detecting pathogenic variants in human genes. Manual preparation of NGS libraries at a large scale is problematic in terms of data quality guarantee and it is also a time-consuming process. To enable high quality (HQ) and high throughput (HT) screening, IAB developed automated WES library preparation using CyBio Felix – Automated Liquid Handling Platform.

Evaluation of quality of WGS and WES results using validated protocols
Whole exome sequencing (WES) and whole genome sequencing (WGS) are standards in human clinical diagnostics and population genomics (POPGEN). We evaluated the quality and genotype concordance of single nucleotide variants (SNVs) and small insertions/deletions (small-indels) in paired blood and saliva genomic DNA (gDNA) isolates for WES and WGS protocols using reference-validated protocols.

From dried blood spot to whole genome long-read sequencing
Dried venous blood spots (DBS) represent a convenient source of gDNA with respect to the small amount of material collected, ease of sample transport and storage. The limitations are mainly related to the difficulties in mapping the short reads to the specific regions of the reference genome and subsequent de novo assembly formation. Long-read sequencing is gaining ground in clinical applications because it overcomes this problem. Illumina Complete Long Reads technology makes long-read sequencing accessible to genomic laboratories by enabling comprehensive human whole genome analysis with both long and short reads data generated in the same sequencing experiment or in two independent experiments at different Illumina sequencing platforms.
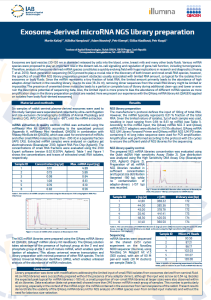
Exosome-derived microRNA NGS library preparation
Exosomes are lipid vesicles (30-150 nm in diameter) released by cells into the blood, urine, breast milk and many other body fluids. Various miRNA species were proposed to play an important role in the distant cell-to-cell signalling and regulation of gene/cell function, including tumorigenesis. Therefore, analysis of exosome-derived miRNA species constitutes a quickly evolving field with huge implications in research and diagnostics (Huang T. et al. 2019). Next-generation sequencing (NGS) proved to play a crucial role in the discovery of both known and novel small RNA species, however, the specifics of small RNA NGS library preparation present obstacles usually associated with limited RNA amount, so typical for the isolates from exosomes or body fluids. Since the miRNA represents a tiny fraction of total RNA, the limited amount primarily leads to the abundance of high adaptor-dimer content in the resulting library. Due to its size (18-34 nt), removing dimer sequences from the small RNA library might be limited or impossible. The presence of unwanted dimer molecules leads to a partial or complete loss of library during additional clean-ups and lower or even ruin the descriptive potential of sequencing data. Also, the limited input is more prone to bias the abundance of different miRNA species as more amplification steps in the library preparation protocol are needed. Here we present our experience with the QIAseq miRNA library kit (QIAGEN) applied on isolates from body fluid-derived exosomes.

Novel long-read sequencing technology in practise
Whole genome short-read sequencing is currently commonly used in number of diagnostic applications. The limitations are mostly related to difficulties in mapping of short reads to the challenging regions of the reference genome such as highly homologous regions and repetitive regions. Long-read sequencing is gaining ground in clinical applications as it overcomes some of these issues. However, many long-read sequencing solutions have been plagued by high DNA input requirements, complex workflows with low throughput and highly variable results and these have limited their utility and adoption. Illumina Complete Long Reads (ILCR) makes long-read sequencing accessible and streamlined for genomic labs. Additionally, this protocol enables generation of comprehensive human genomic data with combination of both long and short reads sequencing.

Published in BMC Genomics (IAB in collaboration with research partners)
Blood-derived genomic DNA (gDNA) is routinely used in the clinical environment. Numerous POPGEN studies and commercial tests benefit from easy saliva sampling. In our validation study we proved, that quality pattern of called variants obtained from genomic-reference-based technical replicates correlates with data calls of paired blood–saliva-derived samples in all levels of tested examinations. When considering non-human-mappable reads, we found that they are present in relatively large proportions in the saliva-derived gDNA when compared with blood-derived gDNA, despite a strict sampling protocol. Although microbiome-to-human misalignment cannot be equivocally ruled out, the results suggest that the end effect does not deviate sequencing accuracy from values typically obtained using blood-derived gDNA.
According to our protocol the saliva can be considered an equivalent material to blood for genetic data analysis.
Kvapilova, K., Misenko, P., Radvanszky, J. et al. Validated WGS and WES protocols proved saliva-derived gDNA as an equivalent to blood-derived gDNA for clinical and population genomic analyses. BMC Genomics 25, 187 (2024).

Adoption of small custom-designed NGS panel for rare FFPE samples of macaque monkey
Whole exome sequencing (WES) is a targeted next-generation sequencing method that identifies protein-coding genes (exons) in the genome. Targeted sequencing using custom-designed NGS panels offers a cost-effective method for detecting rare variants. Important advantage of custom target sequencing is the possibility to personalize the panel (i.e., the inclusion of certain genes and the possibility to sequence exons, specific intronic regions, promoter regions, or the 3′ untranslated regions). On the other hand customized NGS panel designs have to be updated and optimized with regard to gene content or inclusion of known intronic splice variants. It was the optimization of the NGS library preparation from DNA samples using a custom panel that IAB dealt with. The data presented here originate from project of SOTIO Biotech a.s.

Evaluation of WGS and WES protocols based on dried bood spot sample-derived gDNA
Whole exome sequencing (WES) and whole genome sequencing (WGS) have become a standard method in human clinical genetics. Blood-derived gDNA is routinely used in the clinical environment, presenting difficulties associated with invasive sampling. Dried venous blood spots (DBS) represent a convenient gDNA source in respect to low amount of collected material and simple sample transport and storage. However, the use of DBS-derived gDNA for NGS applications has not been analyzed in detail. To address this point, IAB validated both WGS and WES NGS protocols based on DBS-derived isolates.

Validated WGS and WES protocols proved salivaderived gDNA as an equivalent to blood-derived gDNA for clinical and population genomic analyses
Whole exome sequencing (WES) and whole genome sequencing (WGS) have become standard methods in human clinical diagnostics as well as in population genomics (POPGEN). Blood-derived genomic DNA (gDNA) is routinely used in the clinical environment. Conversely, many POPGEN studies and commercial tests benefit from easy saliva sampling. Here, we evaluated the quality of variant call sets, the level of genotype concordance of single nucleotide variants (SNVs), and small insertions and deletions (indels) for WES and WGS using paired blood- and salivaderived gDNA isolates employing genomic reference-based validated protocols.
